A exploração da cor nunca esteve tão evidente como nos dias de hoje, e muitas indústrias são agora direta ou indiretamente dependentes da disponibilidade de corantes artificiais. As indústrias fabricantes e as usuárias de corantes contribuem grandemente para a economia de qualquer país industrializado.
Neste artigo, veremos os tipos de corantes mais comuns disponíveis comercialmente, suas interações com fibras de tecidos e as formas de caracterização de compostos coloridos em função da estrutura molecular, com ênfase nas sínteses e estruturas do índigo, um corante azul do tipo vat muito utilizado no tingimento de tecidos, e do alaranjado de metila, um corante azo utilizado como indicador de pH.
![]()
A
cor tem papel notadamente dominante em nossa vida diária, mas que passa
despercebido na maior parte do tempo. A maior parte da raça humana tem a
habilidade de perceber as cores, e desde a criação da civilização o homem
tenta reproduzir as cores da natureza, tanto por questões estéticas como
puramente funcionais.
Corantes são materiais normalmente aplicados em solução e se fixam de alguma maneira a um substrato, que pode ser um tecido, papel, cabelo, couro ou outros materiais. Idealmente, os corantes devem ser estável à luz e aos processos de lavagem. Também deve apresentar fixação uniforme com as fibras em todo o substrato.
Do ponto de vista comercial, considerável interesse vem sendo demonstrado na avaliação teórica e empírica das relações entre cor e estrutura molecular. Este interesse tem sido acentuado por áreas em expansão onde as cores e os corantes agora adentram, e as relações cor-estrutura são valiosas mesmo para cientistas trabalhando em áreas aparentemente não relacionadas. Sistemas de mostradores de cristal líquido, sensores de radiação de alta energia e impressões coloridas a laser são exemplos recentes dos variados usos da aplicação de corantes sintéticos.
No período anterior à metade do século XIX, os corantes eram quase sempre isolados de fontes naturais, de origem principalmente animal ou vegetal. Naturalmente, as propriedades de muitas destas substâncias estavam longe do ideal e este fato, juntamente com a indisponibilidade comercial das fontes de suprimento, encorajaram a busca por corantes sintéticos com propriedades superiores. O curioso, entretanto, é que o primeiro corante sintético a ser produzido comercialmente foi descoberto por acaso.
Em 1856, William Henry Perkin tentava preparar o alcalóide quinina em seu laboratório, mas seu experimento resultou no isolamento de um corante hidrossolúvel, que forneceu à seda
Descobertas viáveis surgiram rapidamente, e os corantes naturais foram quase que completamente trocados pelos sintéticos no início do século XX. Hoje, virtualmente todos os corantes e pigmentos comerciais disponíveis são substâncias sintéticas, com exceção de alguns pigmentos inorgânicos importantes. Todos os anos centenas de novos compostos coloridos são descritos na literatura, para uma multiplicidade de aplicações.
Os corantes dividem-se nas seguintes categorias principais, de acordo com os processos de tingimento aplicados:
|
Corantes
vat; | |
|
Corantes
diretos; | |
|
Corantes
dispersos; | |
|
Corantes
azo; | |
|
Corantes
trifenilmetilênicos; | |
|
Ftalocianinas. |
Corantes
vat
são exemplificados pelo índigo (figura 1), um composto azul altamente insolúvel
vastamente conhecido desde a antiguidade. Possuía um preço tão elevado, que
apenas a realeza podia compra-lo!
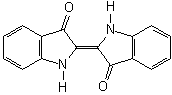
Figura
1 - Índigo
O precursor do índigo, o indoxil, ou 3-hidroxindol, está presente em uma planta de origem indiana, a Indigofera tinctoria, ou na similar européia, Isatis tinctoria. O indoxil é oxidado pelo oxigênio atmosférico ao índigo, provavelmente por dimerização de um intermediário radicalar, o indol:
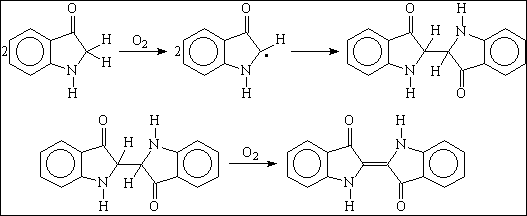
Figura 2:
Reação de oxidação do indol formando o índigo
O índigo é obtido somente na forma trans,pois ocorrem interações do tipo ponte de Van der Walls entre os hidrogênios das aminas e os oxigênios das carbonilas que conferem maior estabilidade à molécula (figura 3). A forma cis desta mesma molécula nunca é obtida porque as interações entre os oxigênios das carbonilas e os hidrogênios das aminas causam repulsão, geando um sistema de maior energia e, portanto, não preferencial e que se converte na forma trans.
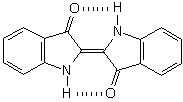
Figura 3
- Estabilização do índigo por interações de Van der Walls
O
índigo foi utilizado no Egito antes do ano 2000 a.C. Na época, era reduzido
por fermentação ao composto leuco, incolor e solúvel. O material a ser tingido era imerso nesta
solução e então exposto ao ar para oxidar a base leuco . Atualmente, o índigo é produzido sinteticamente e reduzido
à forma leuco com ditionita de sódio
(figura 4). Pode ser oxidado pela exposição ao ar ou mais rapidamente pelo uso
de agentes oxidantes como perborato de sódio. O pigmento azul insolúvel
produzido é preso entre as fibras. Esta forma de tingimento era conhecida como vatting,
e deu origem à classificação deste tipo de corante.
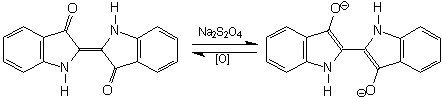
Figura 4
- Conversão do índigo para a forma leuco
com ditionita de sódio
Corantes diretos, como o próprio nome sugere, podem ser aplicados, em solução aquosa, diretamente sobre as fibras. Este processo é especialmente aplicável à lã e à seda. Estas fibras são constituídas por proteínas, que possuem tanto grupos ácidos como básicos que combinam com corantes básicos e ácidos, respectivamente. Um exemplo é a malva (figura 5), o corante que iniciou a síntese industrial de corantes - então conhecido como malveína, mas que atualmente não é mais utilizado.
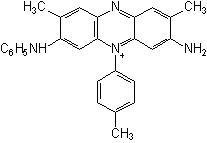
Figura 5
- Malva - inicialmente conhecido como malveína.
Corantes dispersos são aplicados na forma de dispersões aquosas ou suspensões coloidais que formam soluções sólidas com as fibras. Estas fibras não possuem grupos ácidos ou básicos para fixar corantes diretos e são sensíveis à hidrólise nas condições fortemente alcalinas do tingimento vat. Os corantes dispersos apresentam limitações importantes - freqüentemente não apresentam resistência à lavagem (fastness), tendem a sublimar e estão sujeitos a desaparecer com NO2 ou ozônio atmosférico, uma condição conhecida como branqueamento gasoso.
Corantes
azo indubitavelmente constituem a
classe mais importante de substâncias que promovem cor. A versatilidade desta
classe deve-se grandemente à facilidade com que os compostos azo podem ser
sintetizados, e de fato quase todas as aminas aromáticas diazotizadas podem ser
acopladas com qualquer sistema nucleofílico insaturado para fornecer o produto
azo colorido. Se o composto resultante contiver uma amina primária, esta também
pode ser diazotizada e acoplada, fornecendo um sistema de maior conjugação.
Estendendo a conjugação, ou adicionando sistemas cíclicos maiores ou
diferentes grupos doadores de elétrons, uma larga faixa espectral de cores pode
ser obtida, com quase qualquer propriedade física ou química desejável.
Compostos
monoazo insaturados possuem fórmula geral A-N=N-B, onde A e B são sistemas
insaturados cíclicos ou acíclicos conjugados ao grupo azo. Na ausência de
grupos doadores de elétrons, estes compostos são apenas fracamente coloridos,
e a banda de absorção no visível é atribuída à transição de baixa
intensidade pi*
do grupo azo. Se um grupo doador de elétrons é introduzido no ramal A ou B,
uma banda de absorção de alta intensidade é produzida, normalmente na região
do visível, que é normalmente associada à transferência de densidade eletrônica
do grupo doador através de todo o cromóforo. Entretanto, para produzir a máxima
intensidade de transição, normalmente alocam-se todos os grupos doadores de elétrons
no resíduo A, e todos os grupos aceptores de elétrons, se houverem, no resíduo
B. Entretanto, esta não é uma regra geral, e exceções ocorrem.
Corantes azo contendo dois grupos azo são chamados de compostos disazo, e aqueles contendo três grupos azo são conhecidos como trisazo.
Existem
milhares de corantes azo, que podem possuir um ou mais grupamentos azo. Um
exemplo clássico de corantes azo, largamente utilizado em química como
indicador de pH, é o alaranjado de metila:
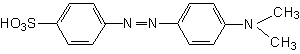
Figura
6 - Alaranjado de
metila.O ácido sulfônico também pode apresentar-se na forma salina.
A síntese de corantes azo requer a diazotização de uma amina aromático para ser acoplada a uma amina ou fenol. Para a síntese do alaranjado de metila, parte-se do ácido sulfanílico e da dimetilanilina.
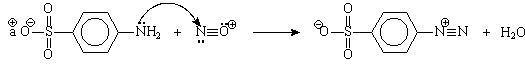
Figura
7:
Reação de formação do ácido sulfanílico diazotizado
O ácido nitroso é formado in situ pela reação de um sal de nitritro inorgânico com ácido clorídrico:
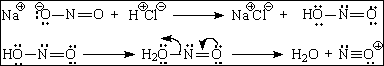
Figura 8 - Preparação do ácido nitroso, que se converte ao íon nitrosônio
em meio ácido
Para que o ácido sulfanílico seja diazotizado faz-se necessária a adição de uma base fraca. Para que a reação de diazotização ocorra a amina deve possuir um par de elétrons livre. Em solução aquosa, o ácido antranílico tende a formar um sal interno, e a adição de base recupera a amina, enquanto forma-se um sal sódico do grupo sulfônico, aumentando a solubilidade do composto.
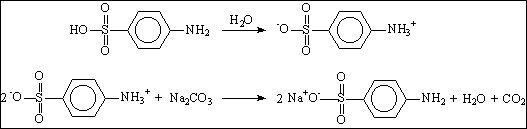
Figura 9
- Disponibilização da amina para reação de diazotização
Para completar a síntese
do alaranjado de metila realiza-se então a reação de acoplamento.
Primeiramente é necessário ativar a dimetilbenzamina que será acoplada ao ácido
sulfanílico diazotizado. Para tanto adiciona-se ao meio reacional uma pequena
quantidade de ácido acético glacial para que a solução mantenha-se levemente
ácida.
Nas reações de acoplamento o controle de pH do meio é fundamental. O reagente eletrofílico é o íon diazônio, ArN2+. Quando o íon hidróxido está presente, o íon diazônio encontra-se em equilíbrio com um composto não ionizado, o diazo-hidróxido ou ácido diazóico, Ar-N=N-OH e, com sais, diazoatos (Ar-N=N-O-Na+) dele derivados:
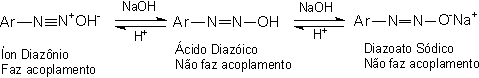
Figura 10
- Formação de ácido diazóico e do respectivo diazoato sódico, que não
fazem reação de acoplamento.
Assim, conclui-se que,
pelo que toca ao agente eletrofílico, a reação de acoplamento é favorecida
por um meio com baixa concentração de íon hidróxido, ou seja, sob pH
ligeiramente ácido.
Entretanto, no que tange ao nucleófilo, duas situações podem ocorrer. Quando o agente de acoplamento é um fenol, soluções ácidas retardam a reação, pois deslocam o equilíbrio ácido para a direita (figura 11). O íon fenóxido reage muito mais rapidamente que o fenol. Em meio ácido, a amina transforma-se no íon respectivo que, devido à carga positiva sobre o nitrogênio, é relativamente pouco reativo para substituição aromática eletrofílica (figura 12). Assim, quanto maior for a concentração de íons hidrogênio, maior será a proporção de amina que se encontra protonada e menor será a velocidade da reação de acoplamento.
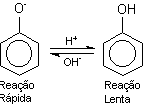
Figura 11
- Equilíbrio iônico dos fenóis
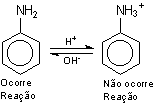
Figura 12
- Equilíbrio iônico das aminas
Assim, reações que envolvem acoplamento de aminas devem constituir um meio-termo. A solução não deve ser demasiadamente alcalina de modo que a concentração do íon diazônio seja baixa, nem tão ácida de tal forma que a concentração de amina livre seja demasiadamente pequena. Verifica-se, na prática, que a reação de acoplamento se produz com maior eficiência em soluções moderadamente ácidas, no caso de aminas, e em soluções moderadamente alcalinas, no caso dos fenóis.
Assim, adicionou-se um ácido fraco, o ácido acético, ao meio para manter o pH da solução em torno de 4 e deslocar o equilíbrio da primeira etapa elementar na figura 19 para a esquerda. Após, o sal de diazônio é adicionado e imediatamente nota-se a formação do precipitado vermelho de heliantina.
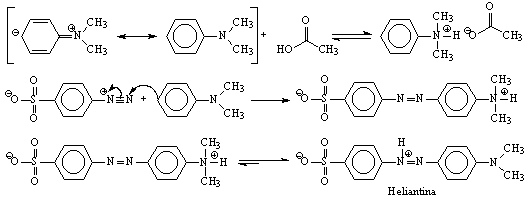
Figura 13:
Reação de acoplamento até a formação do vermelho de heliantina.
Somente quando a heliantina é formada observa-se a coloração vermelha. Enquanto não ocorre a prototropia a deslocalização de elétrons pela molécula é limitada a ponto de não ser suficiente para que a luz promova um salto quântico do orbital HOMO para o LUMO mas, após a formação da heliantina, o efeito de hiper conjugação na molécula permite que tais saltos ocorram, o que confere cor a molécula. A hiper conjugação também é responsável pela estabilização da molécula e, por este motivo, o equilíbrio é deslocado para a formação da heliantina. Após a formação da heliantina, adiciona-se hidróxido de sódio até que o alaranjado de metila se forme:
![]()
Figura 14:
Formação do alaranjado de metila.
Corantes
trifenilmetilênicos
são derivados do cátion trifenilmetílico. São corantes básicos para lã,
seda ou algodão, quando são utilizados mordentes
adequados.
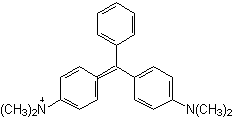
Figura 7
- Verde de malaquita, um corante trifenilmetilênico.
Mordentes são metais de transição que possuem capacidade de se complexar com grupos característicos presentes nas estruturas das fibras, facilitando ou possibilitando a fixação dos corantes. Exemplos de mordentes são os sulfatos de cobre, ferro, alumínio e estanho.
As ftalocianinas são utilizadas como pigmentos ao invés de corantes propriamente ditos. Um membro importante desta classe é a ftalocianina de cobre, um pigmento azul brilhante que pode ser preparado pelo aquecimento de ftalonitrila com íons cobre.
Nem todos os corantes são apropriados para qualquer tipo de tecido. Um corante deve ter a propriedade de fastness - deve ligar-se firmemente às fibras do material e ali permanecer após repetidas lavagens e também não deve clarear quando exposto à luz. Levelness é o termo que se refere à uniformidade do corante na fibra. Um corante level é, assim, aquele que tinge uniformemente o tecido mesmo após sua aplicação.
A forma como um corante se liga a um tecido é um assunto extremamente complexo, uma vez que nem todos os corantes e nem todas as fibras são equivalentes. Um corante bom (fast e level) para lã e seda pode não tingir o algodão, por exemplo.
Para entender os mecanismos pelos quais um corante fixa-se às fibras, deve-se possuir um bom conhecimento das estruturas tanto do corante quanto da fibra. Os principais tipos de fibras são ilustrados na figura abaixo:
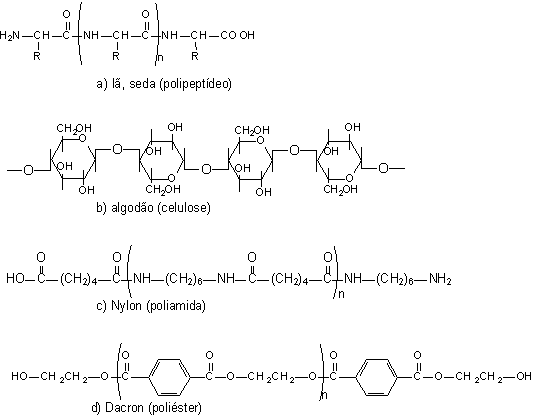
Figura 8
-
Estruturas de fibras de tecidos (a) lã ou seda (polipeptídio), (b) algodão
(celulose), (c) Nylon (poliamida), (d) poliéster. Algodão, lã e seda são
tecidos naturais, enquanto que o nylon e o poliéster são sintéticos.
As duas fibras naturais, lã e seda, são muito semelhantes em suas estruturas. Ambos são constituídos por polipeptídios, isto é, polímeros feitos de unidades de aminoácidos. Estas estruturas possuem um grupo ácido (o ácido carboxílico) e um básico (a amina), e tendem a ligar-se bem com a maioria dos corantes, apesar de exceções ocorrerem, principalmente por efeitos estéricos e/ou geométricos.
Dacron (poliéster), Orlon (acrílico) e algodão (celulose) não contém muitos grupos aniônicos ou catiônicos em suas estruturas e não são facilmente corados por substâncias ácidas ou básicas.
Para interações fibras/corante onde não há possibilidade de ligação do tipo ácido-base, mas que podem formar complexos com metais de transição, muitas vezes são incorporados às fibras íons de metais determinados que fixam o corante por complexação. Entretanto, a escolha deste mordente deve ser realizada com cautela, uma vez que transições metal-ligante podem interferir na coloração final.
Corantes azo fixam-se às fibras que possuem grupos hidroxila por pontes de hidrogênio. Quando o composto é um dizazo, a distância entre os grupos azo deve ser igual, ou muito próxima, a de dois grupos hidroxila na fibra para que o tingimento seja satisfatório. Existe ainda o tingimento in grain, onde primeiramente o tecido é imerso em um banho contendo o sal de diazônio em seguida em outro, contendo o composto aromático nucleofílico ativado. Assim, o corante azo é formado diretamente dentro das fibras dos tecidos.
As origens físicas das cores receberam atenção especial desde o início do estudo sistemático de corantes em 1856 e cedo reconheceu-se que o espectro de absorção de um corante poderia fornecer ao menos uma indicação grosseira de sua cor no uso prático. Durante os primeiros anos do desenvolvimento dos corantes sintéticos, os químicos ficaram intrigados pelas relações existentes entre os espectros de absorção e a estrutura molecular das substâncias químicas, mas antes de 1930 o progresso nos estudos foi severamente impedido pela falta de uma teoria para o processo de absorção de luz. Hoje, graças ao advento da teoria quântica, tratamentos matemáticos com os mais variados níveis de sofisticação estão disponíveis para a predição do espectro de absorção. Igualmente importantes são os tratamentos qualitativos da absorção de luz, estimados pela teoria da ligação de valência e pelas teorias de orbital molecular, que podem ser utilizadas para predizer qualitativamente os efeitos que as mudanças estruturais podem causar no espectro de absorção de uma molécula.
Quando um material interage com a radiação eletromagnética, uma série de processos pode ocorrer, como dispersão, absorção, fluorescência/fosforescência e reação fotoquímica. Em geral, quando utiliza-se radiação na faixa do uv-visível (180-850nm) mede-se a absorção da radiação pelas moléculas dos compostos químicos.
Os espectros de uv-visível geralmente apresentam apenas algumas bandas de absorção largas. Comparadas com técnicas como spectroscopia de infravermelho que produz muitas bandas, a espectroscopia uv-visível fornece apenas poucas informações qualitativas para identificação e caracterização de compostos, mas trata-se de técnica extremamente útil no estudo de substâncias que promovem cor.
Para
comparação da cor observada com o espectro obtido, utiliza-se a correlação
entre a cor absorvida. A cor observada é a complementar da absorvida, conforme
tabela abaixo:
Tabela 1 - Absorbâncias e cores complementares
|
Intervalo de Comprimento de Onda (nm) |
Cor Absorvida |
Cor
Complementar
(ou observada) |
|
650
- 780 |
Vermelho |
Azul
esverdeado |
|
595
- 650 |
Laranja |
Verde
azulado |
|
560
- 595 |
Amarelo-verde |
Roxo |
|
500
- 560 |
Verde |
Roxo-vermelho |
|
490
- 500 |
Verde
azulado |
Vermelho |
|
480
- 490 |
Azul
esverdeado |
Laranja |
|
435
- 480 |
Azul |
Amarelo |
|
380
- 435 |
Violeta |
Amarelo-verde |
O espectro eletrônico tem a propriedade de revelar a energia exata necessária para promover um elétron do HOMO (orbital molecular ocupado de maior energia) para o LUMO (orbital molecular ocupado de menor energia) de uma molécula. Enquanto este elétron está no LUMO diz-se que o composto está excitado, enquanto que quando este elétron retorna para o seu estado fundamental libera exatamente a mesma energia que foi absorvida. Esta energia, quando na região visível do espectro eletromagnético, fornece a cor.
Algumas substâncias absorvem energia em faixas mais baixas de comprimentos de onda, de 200 a 400nm (ultravioleta), mas outras absorvem em comprimentos de ondas mais altos, de 400 a 800nm (visível). Os corantes são substâncias que absorvem energia de comprimentos de onda na faixa do visível. As transições que ocorrem na região do visível são de mais baixa energia que aquelas que ocorrem na região do ultravioleta. Assim, substâncias que possuem um grande número de conjugações e que permitem a deslocalização de elétrons por toda sua estrutura, em geral, possuem cor.
As transições na faixa do visível são de menor energia porque o HOMO e o LUMO estão mais próximos um do outro. São justamente as conjugações que provocam esta diminuição na distância entre o HOMO e o LUMO, pois a deslocalização e carga diminui a energia do sistema. Portanto, apesar da absorção da radiação uv-visível resultar em uma transição eletrônica, a absorção de comprimentos de onda característicos por uma molécula é determinada por seus grupos funcionais - doadores ou retiradores de elétrons - e da capacidade de deslocalização de carga.
A. Krell